I recommend downloading VMWare and Ubuntu and running the operating system virtually. You may need a virtual drive mounting program, which will simulate a physical installation CD, so that you can install Ubuntu. I run Ubuntu on my PC using VMware to run a virtual machine with Ubuntu.
Also, if you will be logging on to a remote server via ssh often you will have to go through some sort of secure logon (for most Universities). I have to use Cisco VPN client to logon. SSH is a "secure shell," which will allow you to log on to another machine and run programs, etc. You will need the username and IP address of the computer to logon remotely. The command to log on will look like: ssh username@11.111.111.111. You will be prompted for a password to that computer. If you are really uncomfortable using text commands, you can always enable Screen Sharing, but this has its downsides. If you have a set of tasks to do in a routine manner, you can write out the commands in advance, in a script, and execute them. With the normal user interface, you'd have to click icons and click menus for certain options. You will not have a record of what you've done (this is why people prefer to use the R software environment as well), and sometimes the repetitive icon/menu clicking is annoying. Automating tasks and scripting are your friend!
Set up a BLAST database:
It is necessary to become comfortable navigating your file folder system using text commands in the Terminal. At first this is difficult for Window's users that are used to a graphical interface. But you can explore using commands like
cd Desktop
or
cd ..
to move up or down in the folder hierarchy.
The command ls is like a flashlight, allowing you to see whats in a folder. In addition to ls, you can use ls -l to see more information about the files in a folder, such as date created, size, what permissions are set, etc. If you want to see hidden files (such as system files that begin with a . ) you can type ls -a.

makeblastdb -in infile.txt -dbtype nucl -out databasename
Here's the general structure of the command:
program -in nameoffile -nextoption option -nextoption option
If I type ssh username@11.111.111.111, I can log on my computer at school, from home, and execute commands remotely. As some of my genome files are 30 gigabytes, this is very convenient. Some of the temporary folders created with bioinformatics programs generate 500 gigabytes of data! Trying to move folders that large, even on a 1 terabyte removable harddrive is a real pain and it eats up a lot of time.
You can use mkdir to make a new folder.
mkdir gene_run
cd gene_run
Now I am in the folder where I want my files to go. Since I am in this folder, when I execute the makeblastdb command and blastn commands, files will be created and the programs will run fine.
NOTE: that if you exit the folder or start in a default folder like Users/name, your files will be created there. This causes confusion for new users, who might try to execute a command without being in the right directory. You might wonder why your files disappeared because you thought you were in the Desktop folder, but weren't.
"WHY WON'T IT WORK? IT DID BEFORE" might be what you ask yourself. Check what folder you are in and be cognizant that your location is implicitly defined when you are using the terminal, but that your location can be critical to running certain programs. When doing BLASTS, you must be in the same folder where you made your database. If you leave the folder, you won't be able to run that BLAST unless you make a new database, etc.
In Windows, to run a program, you click on an icon. In Terminal, you just type the name, followed by the options you want to use to run it. The "program" is makeblastdb and I can tell it what file to use as input, what kind of database I want to make, and the name of the output file. Just separate them by spaces. It doesn't matter what order you put the options in either.
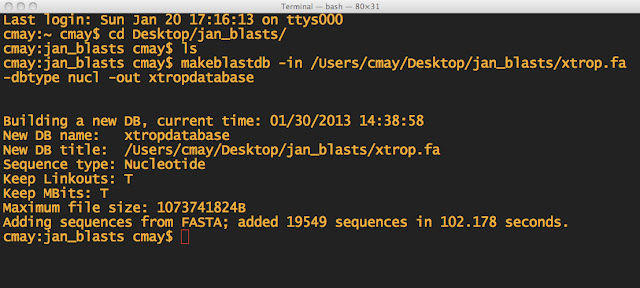
NOTE: First I made sure I was in the directory, which I named jan_blasts. I also made sure the .fa file was in the same folder. THEN I made the BLAST database.
There will be 3 files generated in the directory:
xtropdatabase.nhr
xtropdatabase.nin
xtropdatabase.nsq
These are files that the BLAST program will use.
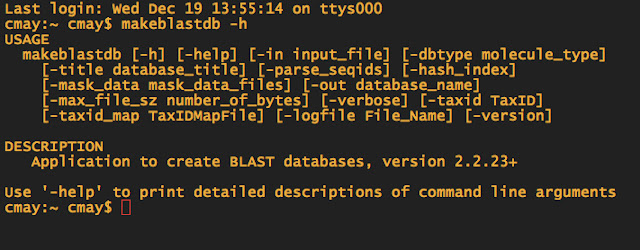
When in doubt about what commands you need to use for doing a BLAST, you can always type makeblastdb -h to pull up the help file.
-dbtype is the database type. Since my Ensembl FASTA file has nucleotide sequences, I choose nucl as the option. If I had proteins, I would use prot etc. -in is the infile -out is the name of your "database" now and you will need to remember the name, for the next step.
When running multiple blasts, which is often the case, you can press the up button at the terminal's blinking cursor and the last command will appear. For doing multiple blasts, this is a good shortcut. You can also drag a file from the desktop to the terminal window and the file path will appear at the blinking cursor.? ?
The input file should be from Ensembl; the gene you want to look at. The output file can be a tabular format for Excel, as denoted by the -m 9. You should look into other output formats, like the 7 option. The -a 4 is instructing the blast to use 4 cores, to speed up the blast.
Getting Gene Sequences
To begin using bioinformatic tools with a Linux-like terminal, proper organization is a must. Because you will be working with a command line, all the files, file names and folders should be structured and arranged very clearly. Otherwise when you have hundreds of files, it gets confusing especially if you move files or archive them. It is advantageous to use one folder for many types of files, although this can be confusing. I have named all my files to include pertinent information (i.e. species_gene_cDNA_hits_extracted.txt), so that if they are moved to another folder, they can easily be identified. This will prevent confusion in the future if files are moved around.
To set up for the blast you should download your gene files from Ensembl, perhaps cDNA, or the exon, depending on what you want. You can export your genes from Ensembl, or just copy the text into a text file. Go to Ensembl, type in a gene and select your species. Then, you will choose the Export Data button on the left sidebar.
cd Desktop
or
cd ..
to move up or down in the folder hierarchy.
The command ls is like a flashlight, allowing you to see whats in a folder. In addition to ls, you can use ls -l to see more information about the files in a folder, such as date created, size, what permissions are set, etc. If you want to see hidden files (such as system files that begin with a . ) you can type ls -a.

makeblastdb -in infile.txt -dbtype nucl -out databasename
Here's the general structure of the command:
program -in nameoffile -nextoption option -nextoption option
If I type ssh username@11.111.111.111, I can log on my computer at school, from home, and execute commands remotely. As some of my genome files are 30 gigabytes, this is very convenient. Some of the temporary folders created with bioinformatics programs generate 500 gigabytes of data! Trying to move folders that large, even on a 1 terabyte removable harddrive is a real pain and it eats up a lot of time.
You can use mkdir to make a new folder.
mkdir gene_run
cd gene_run
Now I am in the folder where I want my files to go. Since I am in this folder, when I execute the makeblastdb command and blastn commands, files will be created and the programs will run fine.
NOTE: that if you exit the folder or start in a default folder like Users/name, your files will be created there. This causes confusion for new users, who might try to execute a command without being in the right directory. You might wonder why your files disappeared because you thought you were in the Desktop folder, but weren't.
"WHY WON'T IT WORK? IT DID BEFORE" might be what you ask yourself. Check what folder you are in and be cognizant that your location is implicitly defined when you are using the terminal, but that your location can be critical to running certain programs. When doing BLASTS, you must be in the same folder where you made your database. If you leave the folder, you won't be able to run that BLAST unless you make a new database, etc.
In Windows, to run a program, you click on an icon. In Terminal, you just type the name, followed by the options you want to use to run it. The "program" is makeblastdb and I can tell it what file to use as input, what kind of database I want to make, and the name of the output file. Just separate them by spaces. It doesn't matter what order you put the options in either.

NOTE: First I made sure I was in the directory, which I named jan_blasts. I also made sure the .fa file was in the same folder. THEN I made the BLAST database.
There will be 3 files generated in the directory:
xtropdatabase.nhr
xtropdatabase.nin
xtropdatabase.nsq
These are files that the BLAST program will use.
When in doubt about what commands you need to use for doing a BLAST, you can always type makeblastdb -h to pull up the help file.

-dbtype is the database type. Since my Ensembl FASTA file has nucleotide sequences, I choose nucl as the option. If I had proteins, I would use prot etc. -in is the infile -out is the name of your "database" now and you will need to remember the name, for the next step.
When running multiple blasts, which is often the case, you can press the up button at the terminal's blinking cursor and the last command will appear. For doing multiple blasts, this is a good shortcut. You can also drag a file from the desktop to the terminal window and the file path will appear at the blinking cursor.? ?
The input file should be from Ensembl; the gene you want to look at. The output file can be a tabular format for Excel, as denoted by the -m 9. You should look into other output formats, like the 7 option. The -a 4 is instructing the blast to use 4 cores, to speed up the blast.
Getting Gene Sequences
To begin using bioinformatic tools with a Linux-like terminal, proper organization is a must. Because you will be working with a command line, all the files, file names and folders should be structured and arranged very clearly. Otherwise when you have hundreds of files, it gets confusing especially if you move files or archive them. It is advantageous to use one folder for many types of files, although this can be confusing. I have named all my files to include pertinent information (i.e. species_gene_cDNA_hits_extracted.txt), so that if they are moved to another folder, they can easily be identified. This will prevent confusion in the future if files are moved around.
To set up for the blast you should download your gene files from Ensembl, perhaps cDNA, or the exon, depending on what you want. You can export your genes from Ensembl, or just copy the text into a text file. Go to Ensembl, type in a gene and select your species. Then, you will choose the Export Data button on the left sidebar.

Next, chose what parts of the gene you want and click next.

You can just copy and paste this text into a text file and name it species_gene.txt. You only need to copy the part until the line break. Subsequent text after the line break will represent other isoforms, and at the end is a long scaffold. To find a gene ortholog in another species, you can just use the first isoform.

You will need to use different commands for nucleotide, protein, etc. The description for tblastn, tblastx ,blastp can be found at Ensembl's Blast Program Selection Guide. The website is well documented, so make use of it. You can download whole genomes from Ensembl from the Downloads section.
Okay, to start the BLAST: You will need to format or make the blast database. Essentially you are designating a file as the “target” which you will be searching a “query” with. The target file is the genome you are using as a reference, and the query is perhaps the individual gene you want to search the target for. i.e. If I want to find A apletophallus genes that are related to development, but I only have the complete genome for A carolinesis to guide me, I would set up the apletophallus contigs I have as the target, to pull out known carolinesis genes, using aapl.fa. Then while blasting, I would use acar_lfng_cnda.txt to search against the aapl database.
BLAST COMMANDS - WHAT ARE THEY?
blastx -help will bring up the command line arguments (or blastn, etc)
After –d you can drag and drop the name of your target database (Acar-contigs1.fa) into the terminal window. You can erase the path name, leaving only Acar-contigs1.fa. The program knows where to look for your target database, because you set your path variables already. Put the path name of your query file after –i, then for –o name a new output file name where you will get your result. I like the m -9 result (also by including a .xls at the end), which is in tabular form and can be opened in excel(appl_lfng_exons_out.xls).
Without the m -9 option, a text file will be created.
Once the blast results have been obtained, I selected the top hits with the greatest accuracy. You should look for a very low e-value, sometimes 10^-50, but ideally over 10^-6. The option is -evalue .00000000001, for instance I then copied the kmer names and created a .txt file for it. (Creating file names with information about the file is very important, once you get a folder with hundreds of files in it!)
To do the BLAST type:
blastn –query acar_gene_dkk2.txt -db Acar-contigs1 -evalue .000000000001 -out acar_results.txt

-db is the name of the database that you specified earlier. BLAST will use this database (the genome file) to search the gene (text query file) against. -evalue should be 10^-10 or so. blastn will do a search using a nucleotide query and a nucleotide database. You may have to use other programs like blastn, blastx, blastp, tblastn, etc. Look on Ensembl's website for an explanation of these programs and when its best to use them. Or you can always check by typing
blastn -h
blastx -h
blastp -h
tblastx -h
A description will appear at the bottom as a reminder of what the program is for.
blastn - Nucleotide-Nucleotide BLAST
blastx - Translated Query-Protein Subject BLAST
blastp - Protein-Protein BLAST
tblastx - Translated Query-Translated Subject BLAST
I'm not going to get into why using a protein query is sometimes the best thing, but there are very good reasons sometimes to do a BLAST using protein sequences. Remember that genes have a lot of non-coding elements, introns are more poorly conserved, and if you are searching for genes in two species that have a long time period say Chicken vs. Stickelback, you might not find a lot of conservation and you could get low hits. However, the its easier to find orthologous matches for protein-coding regions, because proteins have a greater propensity to be conserved for functional reasons.
Grepping
You will also need to know how to grab parts of a sequence from the very large fasta files. With a normal size file, you could just do a search for the text, but this becomes difficult when working with a whole genome. For this reason, grep is your friend! Let's say you get back your BLAST results and you have a match. The file shows you the sequence that matches your query (called the subject, abbreviated sbjct). I copy the sequence.

Next, I use the grep command to pull the sequence from the fasta file and output it to a file on my desktop.
grep -C 15 "sequencehere" inputfile > outputfile

This will give me a text file with the sequence in it, as well as 15 lines above and below my search target, which is good if you want to check out the additional sequence around the exon you are looking at.

Likewise, if you type:
grep -A 3 "sequencehere" inputfile > outputfile

You will get 3 lines returned, after your search target.

Grep -B will print x number of lines before your search target.
grep -B 3 "sequencehere" inputfile > outputfile
Grep can be a great tool to use! Read more here.
Other things
I next created a folder called Tools, with all my perl scripts and important files. In order to access these from the terminal, permissions must be set correctly. In the directory of the file type:
ls –l
This tells you what the permissions are: read-write-execute.
chmod 777 *.pl extract2align.pl
This sets full access to read, write, etc for all files that end in .pl. I set my permissions to full, but you can set them according to your preferences, from 7-0, with 0 locking the file (be careful not to lock yourself out of the file.) If you are working on Saguaro, be aware that others can access your files if you do not restrict access. For more info on the User – Group structure of permissions, see Google. Google is your friend and there are many sites that will have answers about linux or bash that you will need to use as a resource or to solve problems you encounter.
Often times you will want to reference an earlier terminal session to see where you left off. To review terminal history, open the hidden system file in the home folder (username) by:
open .bash_history
To search use command F I keep copies of the .bash_history to keep track of what I've done, as I think this is a good scientific habit for noting bioinformatic methods.
Installing the BLAST binaries can be difficult if you don't have the right software dependencies installed already. At minimum, you should install PERL and make sure it is in your path file in your .bash_profile.
You can type emacs ~/.bash_profile. Add the path to your program and then type Control+X Control+C to save and exit. I have added a long list of my programs to my bash profile.

Emacs is a text-like editor in Mac. You can use it to edit the file, which tells your computer where to look for programs, so that when you type in a program name from any location, it will work. (type which program to find where things are kept. ie. which blastn or which perl will show you the path to where the file is kept. If you want to know where you are you can type pwd and it will print your current location.) Control+Z will let you exit out of Emacs back into the terminal.
To install BLAST software, follow the install instructions, you probably have to tar -xvf emsemble.tar to unpack it into your directory.
Next, I used the perl script to search through the Acar-contigs1.fa to pull out the information on the kmers I want out of this massive file, which would take a long time to search through otherwise. Learn how to use scripts!! They will make your life so much easier. There are plenty of perl scripts that can be downloaded for free from the internet. Make use of as many resources as possible. This extract2align.pl script is attached to this post.
/Users/username/Tools/extract2align.pl --infile /Users/username/Aapl_JanProject/blastdb/ Acar-contigs1.fa --idlist /Users/username/Aapl_JanProject/aapl_lfng_hit.txt --outfile /Users/username/Aapl_JanProject/aapl_lfng_hit_extracted.txt
This executes the extract2align.pl perl script that will dig through my target database and pull out the kmers that were the top hits. It will create a new file with (put the _extracted in the file name, to remain organized) the nucleotides. You can find a copy of this Perl script here.
aapl_lfng_hit_extracted.txt
Results ls –l
This tells you what the permissions are: read-write-execute.
chmod 777 *.pl extract2align.pl
This sets full access to read, write, etc for all files that end in .pl. I set my permissions to full, but you can set them according to your preferences, from 7-0, with 0 locking the file (be careful not to lock yourself out of the file.) If you are working on Saguaro, be aware that others can access your files if you do not restrict access. For more info on the User – Group structure of permissions, see Google. Google is your friend and there are many sites that will have answers about linux or bash that you will need to use as a resource or to solve problems you encounter.
Often times you will want to reference an earlier terminal session to see where you left off. To review terminal history, open the hidden system file in the home folder (username) by:
open .bash_history
To search use command F I keep copies of the .bash_history to keep track of what I've done, as I think this is a good scientific habit for noting bioinformatic methods.
Installing the BLAST binaries can be difficult if you don't have the right software dependencies installed already. At minimum, you should install PERL and make sure it is in your path file in your .bash_profile.
You can type emacs ~/.bash_profile. Add the path to your program and then type Control+X Control+C to save and exit. I have added a long list of my programs to my bash profile.

Emacs is a text-like editor in Mac. You can use it to edit the file, which tells your computer where to look for programs, so that when you type in a program name from any location, it will work. (type which program to find where things are kept. ie. which blastn or which perl will show you the path to where the file is kept. If you want to know where you are you can type pwd and it will print your current location.) Control+Z will let you exit out of Emacs back into the terminal.
To install BLAST software, follow the install instructions, you probably have to tar -xvf emsemble.tar to unpack it into your directory.
Next, I used the perl script to search through the Acar-contigs1.fa to pull out the information on the kmers I want out of this massive file, which would take a long time to search through otherwise. Learn how to use scripts!! They will make your life so much easier. There are plenty of perl scripts that can be downloaded for free from the internet. Make use of as many resources as possible. This extract2align.pl script is attached to this post.
/Users/username/Tools/extract2align.pl --infile /Users/username/Aapl_JanProject/blastdb/ Acar-contigs1.fa --idlist /Users/username/Aapl_JanProject/aapl_lfng_hit.txt --outfile /Users/username/Aapl_JanProject/aapl_lfng_hit_extracted.txt
This executes the extract2align.pl perl script that will dig through my target database and pull out the kmers that were the top hits. It will create a new file with (put the _extracted in the file name, to remain organized) the nucleotides. You can find a copy of this Perl script here.
aapl_lfng_hit_extracted.txt
hes1_aminos_hits.txt
--------------------
k31:121332
k51:3345759
------------------------------
>k31:121332
ACACGACTACAGCTCCTCGGACAGCGAGCTAGACGAGAACATCGAGGTGGA
GAAGGAGAGTGCAGACGAGAATGGAAACCTGAGTTCAGTGCCGGGGTCCAT
GTCCCCCTCCACGTCCTCCCAGATCTTGGCCAGAAAAAGGCGCAGAGGGGT
GATCGAGAAACGCCGGCGAGATCGAATCAACAACAGTTTATCTGAATTAAG
GAGACTGGTGCCCAGCGCTTTCGAGAAACAGGGATCGGCGAAGCTGGAAA
AGGCAGAGATTTTGCAAATGACCGTAGACCACCTGAAAATGCTGCACACGG
CAGGAGGAAAGGGGTACTTCGACGCTCACGCTCTGGCGATGGACTACCGCAG
CCTAGGGTTCCGGGAGTGCCTGGCAGAGGTAGCACGGTACCTCAGCATCATC
GAGGGCCTGGACACCTCCGACCCT
------------------------------
(or if the .fa was run on only one contig, there will be no k31 before the subject id number. Just use the subject id number that came up in your _out.xls file.)
If you did –m 9 for the tabulated file, your excel file will not have any column information, so copy and paste this into the first row of your excel spreadsheet.
Query Subject ID % ID length mismatches gaps query start query end subject start subject end e value bit score
You want to look for both length and e-value to determine which is most likely the gene match you are looking for.
Next I use Geneious and drag the _extracted file into the window. Next you can do alignments with another genome/gene to compare. You can also use UCSC'S Genome Browser.
(or if the .fa was run on only one contig, there will be no k31 before the subject id number. Just use the subject id number that came up in your _out.xls file.)
If you did –m 9 for the tabulated file, your excel file will not have any column information, so copy and paste this into the first row of your excel spreadsheet.
Query Subject ID % ID length mismatches gaps query start query end subject start subject end e value bit score
You want to look for both length and e-value to determine which is most likely the gene match you are looking for.
Next I use Geneious and drag the _extracted file into the window. Next you can do alignments with another genome/gene to compare. You can also use UCSC'S Genome Browser.
Sorry about the font color problems. I tried to make all the font black but there is an error with the Blogger editing page that won't let me change it.
ReplyDelete